Research interests :
I am interested in the developpement of new computational methods to study genome evolution and dynamics. At the moment, my interests are focused on methods dedicated to sequencing data, Next and Third generation sequencing technologies. I develop methods for de novo genome assembly, genomic variant detection and analysis, with a particular focus on Structural Variation.
In the past, I worked on classical comparative genomics methods based on the identification of homologous genes (Post-doc work on Mollicutes) and whole genome comparisons (PhD work).
From a biological point of view, I am particularly interested in genome rearrangements and genome organisation, such as the analysis of rearrangement breakpoints in complete genomes (PhD work) or the detection of structural variants in population genomics data (current work).
Outline :
- New methods for sequence and genome comparison with sequencing data (2011 - now)
- Structural variant genotyping with long reads
- Large insertion detection and assembly with short reads
- De novo variant detection with short reads
- Efficient comparison of raw read sets, application to de novo comparative metagenomics
- Applications to insect genomes : de novo assembly of heterozygous genomes, symbiont analyses, comparative genomics...
- Chromosomal rearrangement breakpoints in mammalian genomes (2005 - 2010)
- Method for precise detection of rearrangement breakpoints: Cassis
- Model of chromosomal evolution
- XY inversions
- Spatial synteny
- Comparative genomics of Mollicutes (Post-doc, 2008 - 2010)
New methods for genome comparison with sequencing data (2011 - now)
Genomics studies entered an unprecedented deep change with the arrival of Next Generation Sequencers (NGS), and now Third Generation Sequencers (TGS). These new technologies enable to sequence biological material with a flow much higher than before, for a price now accessible to most biological lab. However these generate huge amounts of data of a new type (mainly short sequences called reads) that necessitate new computational methods.
Notably, some tools that I developed for short read data rely on the Genome Assembly Tool Box (GATB) library which offers a very light representation of the De Bruijn graph. Therefore the time and memory requirements are unprecedently low, enabling for instance to call SNPs for several human datasets on current laptop or desktop computers.
More recently, I focused my research on methods for Structural Variant detection and analysis. Advances in sequencing technologies (long reads) have revealed the prevalence and importance of structural variations (deletions, duplications, inversions or rearrangements of DNA segments) which cover 5 to 10 times more bases in the genome than the point mutations commonly analyzed. I work on methods to improve their detection, genotyping, analysis with various sequencing data types (short, linked and long reads) and in various organisms (human, insects, bacterial symbiont...).
Structural variant genotyping with long reads
One of the problems in Structural Variant (SV) analysis is the genotyping of variants. It consists in estimating the presence or absence of a set of known variants in a newly sequenced individual. We proposed one of the first tool dedicated to SV genotyping with long read data, SVJedi (2020). Then, we extended this method with SVJedi-graph (2023), which uses a sequence graph instead of linear sequences to represent all the alleles of the SVs. We showed that this graph model prevents the bias toward the reference alleles and allows maintaining high genotyping accuracy whatever the proximity of variants.
On the human gold standard HG002 dataset, SVJedi-graph obtained the best performances, genotyping 99.5% of the high confidence SV callset with an accuracy of 95% in less than 30 min.
Software : SVJedi and SVJedi-graph
Large insertion detection and assembly with short reads
We proposed a new method for the integrated detection and assembly of insertion variants from short read re-sequencing data. Importantly, it is designed to call insertions of any size, whether they are novel or duplicated, homozygous or heterozygous in the donor genome. MindTheGap uses an efficient k-mer based method to detect insertion sites in a reference genome, and subsequently assemble them from the complete set of donor reads.
The method is implemented in the tool MindTheGap and showed high recall and precision on simulated datasets of various genome complexities. When applied to real C. elegans and human NA12878 datasets, MindTheGap detected and correctly assembled insertions longer than 1 kb, using at most 14 GB of memory.
MindTheGap has been greatly improved since its publication in 2014. It is also integrated in tools for other applications requiring local assembly, such as targeted assembly of symbiont genome (MinYS) and local assembly of specific loci with linked-read data (MTG-Link).
Publications : MindTheGap : integrated detection and assembly of short and long insertions and Towards a better understanding of the low recall of insertion variants with short-read based variant callers
Software : MindTheGap
De novo variant detection with short reads
We developped tools, such as discoSnp and TakeABreak, to detect genomic variants in raw read sets without assembly nor mapping on a reference genome. The method relies on the exploration of the De Bruijn graph generated from the combined read sets, looking for specific topological motifs. discoSnp focuses on Single Nucleotide Polymorphism (SNP), whereas TakeABreak detects inversion and translocation breakpoints. We developped also efficient filters and ranking to discard false positives in complex and repeated genomes, achieving better results than existing tools or the classical strategy of assembly+mapping. We also extended discoSnp to RAD-seq analyses.
Publication : Reference-free detection of isolated SNPs
Software : discoSnp++ and TakeABreak
Efficient comparison of raw read sets, application to de novo comparative metagenomics
We developped several tools to compare and extract similar reads between raw read sets in an efficient manner. The main focus was on time and memory performances in order to be able to deal with huge metagenomic datasets. This is achieved by using the kmer (word of size k, with k around 20-40) as a comparison unit. Simka computes various pairwise distances between metagenomics datasets based on their common and specific kmer contents. It relies on an efficient kmer counting algorithm and avoids storing the whole kmer count matrice in memory.
Simka was applied notably to compare hundred of sea water samples from the Tara Ocean expedition.
Publication: Multiple comparative metagenomics using multiset k-mer counting.
Software : Simka
More applied projects, mainly with insect genomes : de novo assembly of heterozygous genomes, symbiont analyses, comparative genomics ...
I am also (or was) involved in more applied projects, where we apply our methods (and other classical ones) to original sequencing data and analyse the results.
- Structural variation detection and analysis with linked-read data to study adaptation of a seaweed fly to climatic change, in collaboration with Claire Mérot (CNRS Ecobio, Rennes)
- Pangenome analysis of a complexe of species of alpine butterflies to study hybrid speciation, in collaboration with Laurence Desprès (CNRS LECA, Grenoble)
- Structural variation analysis and local assembly in mimetic butterflies, in collaboration with Mathieu Joron (CNRS CEFE, Montpellier)
- De novo genome assembly of highly heterozygous genomes (tropical butterflies), in collaboration with Marianne Elias (MNHN Paris), ANR Specrep
- Multi-scale characterization of symbiont diversity in the pea aphid complex through metagenomic approaches,in collaboration with Jean Christophe Simon (INRA Rennes).
- Comparison of two strains of the lepidoptera Spodoptera frugiperda, in collaboration with Emmanuelle d'Alençon (INRA Montpellier), ANR Ada-spodo.
- Identification of polymorphism in 40 pea aphid re-sequenced genomes, in collaboration with Jean Christophe Simon (INRA Rennes), ANR SpeciAphid.
Chromosomal rearrangements in mammals
See also my PhD manuscript (in french) and the slides of my defense.
Chromosomal rearrangements are large scale mutations that alter the structure and organisation of genomes. I have studied them in the scope of the evolution of the mammalian genomes. The aim of this work was to characterise the genomic regions which have undergone such events; the latter are called breakpoints.
Cassis
I developped Cassis, a new method to precisely localise rearrangement breakpoints on a genome by comparison with the genome of a related species.
The originality of the method is that it is divided in two steps :- first detecting broadly the synteny blocks;
We proposed a formal definition of synteny blocks between two genomes allowing some flexibility governed by one parameter, together with an algorithm to find them. (Blocks (Ar,Ao) and (Br,Bo) in the picture below) - then refining each breakpoint separately.
The refinement is done by aligning each breakpoint sequence against its specific orthologous sequences in the other species (sequences Sr against SoA and SoB in the picture below). We can then look for weak similarities inside the breakpoint, thus extending the synteny blocks and narrowing the breakpoints. The identification of the narrowed breakpoints relies on a segmentation algorithm and is statistically assessed.
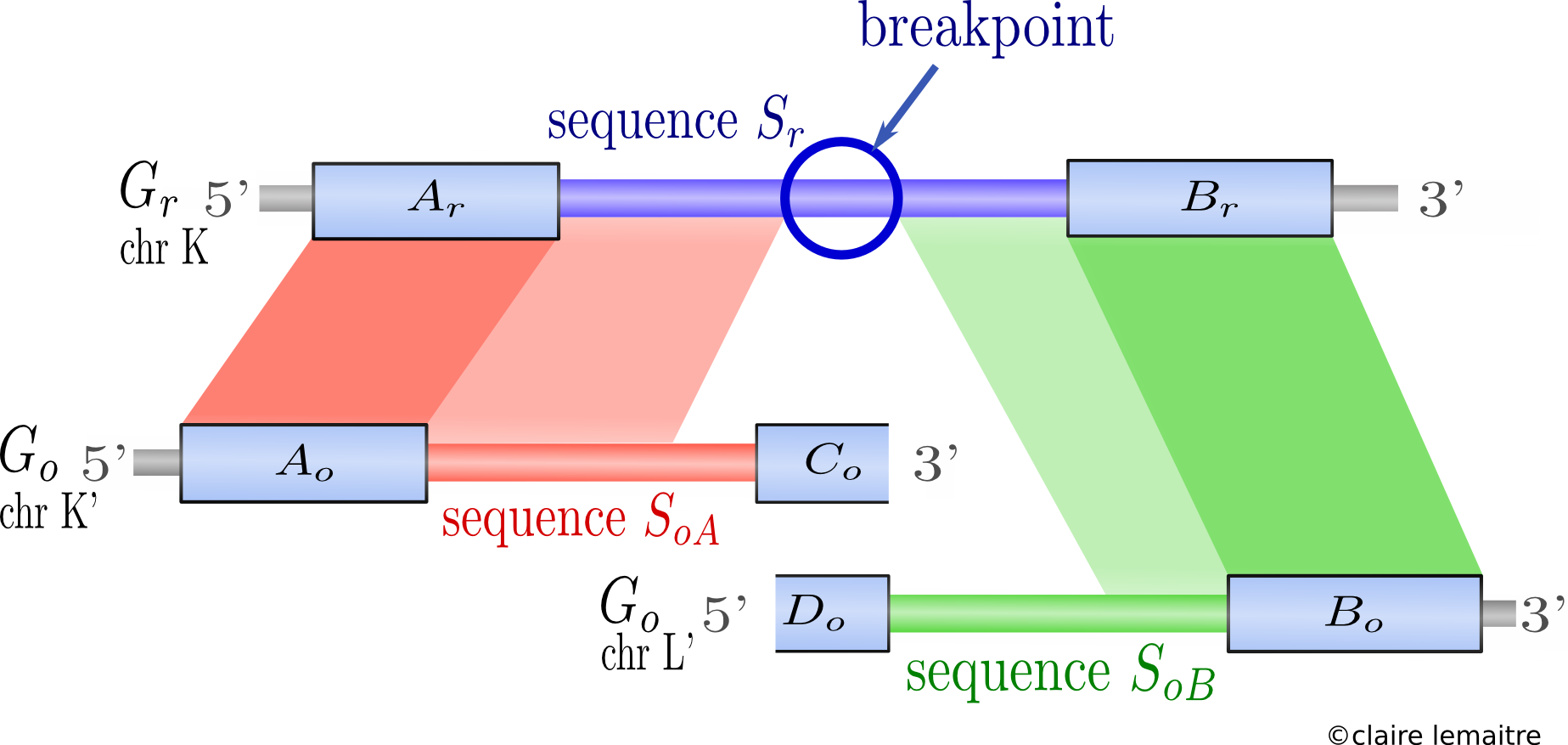
The whole method was applied to localise breakpoints on the human genome compared with other fully sequenced mammalian genomes, and was shown to achieve a better precision than other published methods.
Publications : Precise detection of rearrangement breakpoints in mammalian genomes., Cassis: Detection of genomic rearrangement breakpoints.
Software : Cassis
- first detecting broadly the synteny blocks;
Distribution of rearrangement breakpoints in mammalian genomes
In collaboration with the team of Alain Arnéodo of the Laboratoire Joliot-Curie at the ENS (Lyon), I analysed the distribution of a set of 622 mammalian rearrangement breakpoints (obtained with the method Cassis) along the human chromosomes.
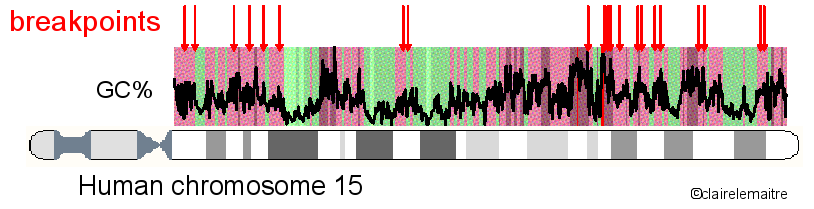
We found that their distribution is highly heterogeneous and follows the organisation of the genome into isochores, with a high density of breakpoints in regions of high GC content and high gene density.
We then proposed the hypothesis that regions of high transcriptional activity that are probably in an open chromatin state, may have an enhanced susceptibility to breakage.Rearrangement and evolution of human X-Y chromosomes
In collaboration with Gabriel Marais of the LBBE laboratory (Lyon), I analysed the rearrangements between human X and Y chromosomes. More precisely, we were interested in the breakpoints at the evolutionary strata boundaries (Y is supposed to have evolved from the X chromosome by several large inversions supressing recombination in the strata defined).
Using the method Cassis, I could identify two sets of duplications clearly linked to the inversions responsible for the formations of stratum 4 and stratum 5. These were not only clear evidence of the existence of these inversions, but also permitted to order them in time (see picture below : the progressive reduction of the PAR (pseudo-autosomal region) by two inversions on the Y chromosome associated with duplications).

Publication : Footprints of inversions at present and past pseudoautosomal boundaries in human sex chromosomes.
Spatial synteny
In this study, we analysed the correlation between 3D chromatin interaction data (public data obtained with the Hi-C method) and breakpoint regions resulting from evolutionary rearrangements in the human genome. We found that two loci distant in the human genome but adjacent in the mouse genome are significantly more often observed in close proximity in the human nucleus than expected. These findings strongly suggest that part of the 3D organisation of chromosomes may be conserved across very large evolutionary distances. To characterise this phenomenon, we proposed to use the notion of spatial synteny which generalises the notion of genomic synteny to the 3D case.Publication : Close 3D proximity of evolutionary breakpoints argues for the notion of spatial synteny.
Comparative genomics of mollicutes
I am involved in the ANR project EvolMyco. The aim of the project is to understand the evolution and adaptation of ruminants mycoplasmas (small bacteria belonging to the mollicutes group). It also includes the sequencing and annotation of 20 new genomes of these species.
Within this project, I focused mainly on the prediction of orthologous relationships between genes. Based on the observation that Mollicute genomes have a strong compositional bias (AT-richness), I proposed a general and simple methodology to build new matrices fitted to specific compositional bias of proteins. I designed such a new matrix MOLLI60 for Mollicute genomes (to replace BLOSUM62) and it enabled to better predict homologous and orthologous relationships thanks to a better estimation of protein alignment significance.
I developped a mixed strategy based on pairwise alignment clustering and detection of species overlap patterns in protein family phylogenetic trees, to predict orthologous relationships between about 60 Mollicute gene sets. The resulting orthologous groups are available in Molligen (a database dedicated to the comparative genomics of Mollicutes, developped by the CBiB) and are currently being analysed in order to find correlations between gene sets and phenotypic traits such as hosts specificities or pathogenicity levels.